Documentation
Outline
- Protein dataset
- Structural motif extraction
- Description of structural words
- Parameters computed on loop dataset:
- Statistic parameters: Occurrence, ORscore
- Geometric parameters: RMSD
- Sequence parameters: Zmax, AALogo
- Parameters computed in SCOP superfamilies: Lpmax, sf*, nbsf*
- Identification of motifs of interest
- Recurrent motifs
- Over-represented motifs in loop dataset
- Ubiquitous motifs
- Functional candidate motifs
- Abbreviations used in the website
1- Protein data set
A list of 5,429 protein structures classified in SCOP was extracted from the PDB using the following criteria: data obtained by X-ray diffraction, with a resolution better than 2.5 Å longer than 30 residues, less than 50% sequence identity between any pair. As it is assumed that proteins grouped in the same SCOP superfamily have similar structure and functions, this level was chosen for our analysis. For statistical analysis, we further restricted the list to proteins classified into superfamilies with at least two members in the data set, corresponding to 4,911 proteins from 1,493 superfamilies. We extracted from this data set 90,811 protein loops.
[ Back to top ]2- Structural motif extraction
To extract structural motifs from loop structures we used the protocol presented in (Regad et al., 2010) . It is based on the simplification of the protein 3D structures into 1D sequences using the structural alphabet HMM-SA (Hidden Markov Model - Structural Alphabet) (Camproux et al., 2004) .
[ Back to top ]Simplification of 3D protein structures using HMM-SA
First, protein structures were described only their α-carbon positions. Each α-carbon backbone was split into overlapping four-residue fragments. The geometry of fragments was defined by a vector of three inter-α-carbon distances and the projection of the last α-carbon on the plan formed by the three others. These resulting distances were the input of a Hidden Markov Model (HMM) in order to class the four-residue fragments according their geometry. It results in an optimal classification, named HMM-SA, with 27 classes grouping four-residue fragments with similar geometry. For each class, labeled by a letter {a,A-Z} and named structural letters (SLs), an average prototype was computed. In addition to the definition of the 27 SLs, HMM allowed also the identification the transition matrix containing the probability of one SL is following by another. Some transitions between SLs are not possible resulting in a limited number of pathways between SLs and short patterns of SLs.
A comparison between the secondary structures and the 27 SLs has shown that four structural-letters [a, A, V, W] specifically describe α-helices, and five SLs [L, M, N, T, X] specifically describe β-strands. The remaining 18 SLs [B, C, D, E, F, G, H, I, J, K, O, P, Q, R, S, U, Y, Z] allow description of loop structures (Camproux et al., 2004) .
The encoding process of a protein structure of n residues consists to search the Maximum A Posteriori (MAP) encoding (SL sequence) using the Viterbi’s algorithm. The input of this algorithm is the vector of 4 geometric descriptors of all four-residue fragments extracted from the protein target. This translation takes into account both the structural similarity of four-residue fragments with the 27 SLs and the preferred transitions between SLs. The output of the encoding process is the sequence of of n-3 SLs, where each SL corresponds to the geometry of a four-residue fragment (Camproux et al., 2004) .
[ Back to top ]Loop extraction
From the SL sequences corresponding to the structure of the 4,911 proteins, we defined secondary structures using a definition based on the SLs (Regad et al., 2006) :
- α-helices are defined as series of at least three α-letters [A,a,V,W]. Only isolated non α-letters are allowed within an helix. Letters [Z,B,C] are allowed at helix termini.
- β-strands are defined as series of at least two &bqeta;-letters [L, M, N, T, X]. Only isolated non β-letters are allowed within a strand. Letters [J,K] are allowed at helix termini.
- Loops are SL sequences linking secondary structures.
From the set of SL sequences, we extracted 90,811 protein loops encoded into HMM-SA.
[ Back to top ]Structural motif extraction
Each the SL sequence of loops were split into structural words, i.e. successive four SL, that correspond to clusters of seven-residues fragments with similar structure (RMSD=0.85 Å) and with amino-acid specificities ( Regad et al., 2010 ). Using this protocol, we extracted, from the 90,811 simplified loops, a total of 25,304 different structural words describing the conformation of 238,158 seven-residue fragments. In (Regad et al., 2010) , we have showed that structural words with a low frequency are linked to structural flexibility and regions with uncertain coordinates. Thus, we did not consider structural words detected less than five times. This resulted in a set of 11,294 words, grouping 224,148 seven-residue fragments.
[ Back to top ]3- Description of structural words
Each structural word was then described by different parameters in order to inform the users on the particularities of structural motifs and to help the users to identify the important regions of protein loops. Firstly, we extracted properties of words computed on loop data set in order to identify non random conserved regions in protein loops constituing the structural redundancy of loops. Then, a second description of words is provided related to SCOP superfamilies, grouping protein domains which have an evolutionary relationship, in order to identify link between structural motifs to protein function.
[ Back to top ]Parameters computed on loop data set
These parameters are used to identify structural motifs conserved across protein loops corresponding to non random regions in loop structures. Three properties are analyzed using this description:
- redundancy of structural words across protein loops
- structural variability of words
- amino-acid (AA) conservation
Statistic parameters
At first, we computed parameters allowing to analyse the redundancy of each word in loop structures.
Occurrence: corresponds to the number of times of a word is seen in the loop dataset, that means the number of seven-residue fragments encoded into this structural word. If a word has an occurrence above 30, it is defined as recurrent word.
ORscore : corresponds to the over-representation score of the word in all loop data-set. This score is obtained after comparing the occurrence of a word in the data set (N) and its expected occurrence (Nexp) computed under a background reference model (an 1-order Markov model for which the parameters are estimated from the global set of loops).
ORscore = -log10[P(Nexp > N)]
when w is seen more than expected
ORscore = +log10[P(Nexp < N)]
when w is seen less than expected
A word with a positive ORscore is defined as over-represented, that means its occurrence in dataset is larger than expected ones. A word with a negative ORscore is defined as under-represented, that means its occurrence in dataset is smaller than expected ones. For example, an ORscore equal to 21.3 means that the word is over-represented with a p-value equal to 10-21.3. A ORscore equal to -17.7 means that the word is under-represented with a p-value equal to 10-17.7.
The ORscore threshold for statistical significance is set to 5.94, using the Bonferroni adjustment to take into account multiple tests. This permits to classify words as over-represented (ORscore > 5.94), under-represented (ORscore < -5.94) or not significant (-5.94≤ORscore ≤5.94).
The computation of ORscore is performed using the SPatt software (Nuel et al., 2010) .
Geometric parameters
RMSD: corresponds to the α-carbon RMSD (Root-Mean-Square Deviation) computed between seven-residue fragments encoded by the same word. This score quantifies the structural variability of a structural word. The smaller RMSd of a word is, the more similar geometry of seven-residue fragments is. This parameter is computed only for recurrent words.
Sequence parameters
These parameters are computed only for recurrent words. These parameters allow the analysis of the sequence-to-structure dependence between words and their amino-acid sequences.
Zmax: This score quantifies the amino-acid specificity of the most significant position among the seven position. For each word, we computed a Z-score for the 20 amino acids for the seven positions resulting in 140 Z-score.
The Z-score of amino acid a, (1 ≤ a ≤ 20 ) at position l (1 ≤ l ≤ 7) of a word w, is obtained by comparing the observed frequency of a at position l in w (Na,l,w) with its expected one (Na,l,wexp):
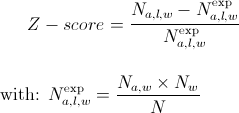
where Nw is the occurrence of w, Na,l is the occurrence of a in position l in all words and N is the total occurrence of all words.
A positive Z-score corresponds to an over-representation of the amino acid, and a negative one corresponds to an under-representation of the amino acid.
Thus, we have 140 Z-score (a Z-score for each amino-acid (20) in each position (7) of a word. To facilitate the analysis of the amino-acid informativity of each word, we computed the maximum Z-score, noted Zmax among the 140 for each words. The higher Zmax of a word is, the stronger amino-acid specificity of the word is.
AALogo :is a logo of the amino-acid sequences of a word. Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position. The Logo is produced by WebLogo.
[ Back to top ]Parameters computed on SCOP superfamilies
This parameters are computed only for words seen more than 5 times. These parameters provide informations about the potential role of the structural word in protein function. To perform that, we combined the over-representation of words and the SCOP classification that is an hierarchical classification of protein domain. At superfamily level, SCOP groups domains according their evolutionary relationships, that means domains with similar structure and function. We suppose that a structural motifs is involved in a functional site, if it is conserved during evolution accross protein containing this functional site (protein grouped in the same superfamily), resulting in an over-representation of the motif in the superfamily. At first, protein loops are classified according to the SCOP accession number of proteins (superfamily level). For each word, we then computed the ORscore in each superfamily (group of SL sequences corresponding to the loops belonging to this superfamily). To facilitate the analysis of the specificity of words, we computed two scores:
Lpmax: corresponds to the maximal ORscore of the word for all superfamilies. This score indicates the highest degree of over-representation for different SCOP superfamilies, that means a quantification of its specificity to the superfamily where is the most specific, named sf*. The more higher Lpmax is, the more specific words is to a superfamily.
nbsf*: corresponds to the number of superfamilies in which a word is significantly over-represented. For example, if a structural word has a Lpmax of 140 and a nbsf* of 3, this structural word is specific to three superfamilies and very strongly for one of them.
[ Back to top ]3- Motifs of interest
[ Back to top ]Recurrent words
These motifs are defined as motifs seen more than 30 times in the loop dataset. These motifs correspond to structures of seven-residues repeated accross loop structures, and shared by different loops.
[ Back to top ]Over-represented words
These motifs are motifs with an ORscore higher than 5.95, that means they are significantly over-represented in the loop dataset. In (Regad et al., 2010) , we have shown that these motifs present particular properties (sequence and structural). Moreover, the over-representation of these motifs, means that they are conserved during the evolution, and thus it seems that these motifs are of interest for proteins.
[ Back to top ]Ubiquitous words
Ubiquitous motifs are defined as motifs over-represented in several superfamilies (with large nbsf∗ (nbsf∗ > 5) These motifs correspond to particular structures of seven-residue fragments shared by different superfamilies. Indeed, they are conserved during the evolution in proteins with different functions It seems that these ubiquitous motifs are not important for the protein function, but are involved in protein structures.
[ Back to top ]Functional candidat words
Functional candidat motifs correspond to motifs highly over-represented, with large Lpmax (Lpmax > 20), in a small number of superfamilies, with weak nbsf∗ (nbsf∗ < 5). These motifs are conserved during evolution in only several superfamilies and they are thus strongly specific only to proteins with the same function. So, it seems to they have a functional role in proteins.
[ Back to top ]